

Orchard Therapeutics今天宣布,美国FDA已接受其基因疗法Libmeldy(atidarsagene autotemcel)治疗异染性脑白质营养不良(MLD)的生物制品许可申请(BLA),并授予优先审评资格。FDA预计于2024年3月18日前完成审查。试验数据表明,施以该一次性基因疗法能够持续保留患者的运动功能和认知发育能力。
MLD是一种罕见且危及生命的机体代谢系统遗传性疾病,根据现有文献估计发生率约为1/10万新生儿。MLD是由芳基硫酸酯酶-a(ARSA)基因突变引起的,导致硫酸盐在脑和身体其他区域蓄积,包括肝脏、胆囊、肾脏和/或脾脏。随着时间的推移,患者的神经系统会受损,导致其在运动、行为和认知上退化,并发生严重痉挛和癫痫发作等神经问题。MLD患者会逐渐丧失活动、说话、吞咽、进食和视物的能力。在婴儿晚期,发病后5年的死亡率估计为50%,10年的死亡率估计为44%。
Libmeldy(又名OTL-200)使用慢病毒载体将编码芳基硫酸酯酶-A的ARSA转基因导入到患者自体CD34阳性造血干细胞和祖细胞中,该疗法已在欧盟获批用于治疗以ARSA基因双等位基因突变导致儿童ARSA酶活性降低为特征的MLD患者。Libmeldy是首个获批用于符合资格的早发性MLD患者的疗法。
该BLA申请是基于39例早发性MLD儿科患者的数据,这些患者入组了两项前瞻性非随机临床研究(n=30)或在扩大使用框架下接受治疗(n=9),给予Libmeldy并与49例未接受治疗患者的自然史数据进行比较。在临床试验中,与疾病自然史相比,Libmeldy治疗可使大多数患者的运动功能和认知发育得以保留,随访时间长达12年(中位数6.76年)。
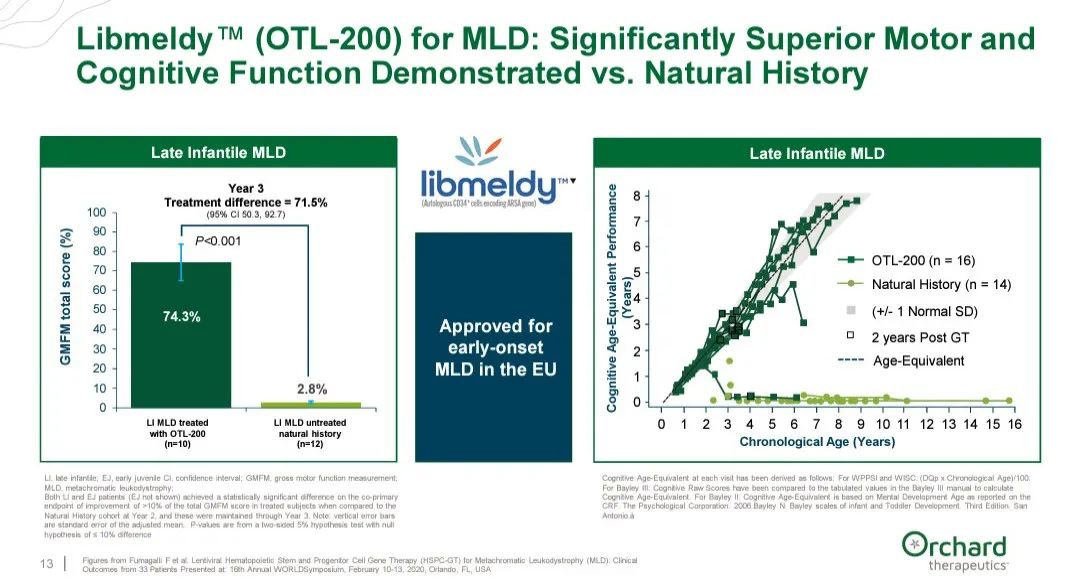
▲Libmeldy试验结果(图片来源:参考资料[2])
累积随访数据显示,Libmeldy治疗通常显示良好的耐受性,无治疗相关严重不良事件或死亡。大多数不良事件与白消安(busulfan)预处理或背景疾病相关。
关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..四川省医药保化品质量管理协会举办2026
当前,制药行业正处于合规升级与绿..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..四川省医药保化品质量管理协会党支部召
按照省市场监督管理局社会组织联合..


