

▎药明康德内容团队编辑
1988年“快速通道”认定出台之后,FDA陆续出台了一系列加快审评审批计划。20年来,获得FDA批准的新药和生物制品,新药获批所依据的关键性临床试验证据(pivotal clinical evidence),呈现出什么样的变化发展趋势?
1988年,FDA公布加速针对无药可治的患者的新治疗药品审评,被视为“快速通道”的开端。20年来,快速通道资格和随后推出的一系列加快审评审批计划,在加速新药与生物制品上市的同时,对FDA药品审评审批所依据的关键性临床试验(pivotal trial),以及新药上市后的确证性临床试验,提出了相应要求。
关键性临床试验,指证明新药疗效,作为监管机构批准依据的临床试验。关键性临床试验通常为3期临床研究。上世纪80年代末,“快速通道”出台后,FDA曾经以行业指南的形式,要求开展至少两项关键性临床试验。但是,关键性试验并非只限于3期试验,对于某些获批药品,2期试验也可被视为关键性试验。例如,对于罕见病治疗药物,从明智监管的角度看,一刀切地要求开展两项关键性临床试验,在很多情况下并无必要。
近日,耶鲁大学医学院Joseph S. Ross医生等,在JAMA Network Open上发表论文,详细分析了20年来FDA批准的新药与生物制品所依据的有效性的临床证据变化发展趋势。
这项研究,选取了1995-1997年,2005-2007年,以及2015-2017年间获得FDA批准的针对339种适应症的273个新药和生物制品。
根据产品类型、治疗领域,孤儿药资格,以及所采用的具体审评审批通道(例如优先审评、加速批准)对调研的治疗药物分类。研究所调研的关键性临床试验特征包括是否使用随机化,置盲,对照类型(comparators),主要终点,所治疗的患者数,以及临床试验时间。
▲表1. 1995-1997年,2005-2007年和2015-2017年间FDA批准新药与生物制品和相关适应症情况(数据来源:参考资料[2],药明康德内容团队制图)
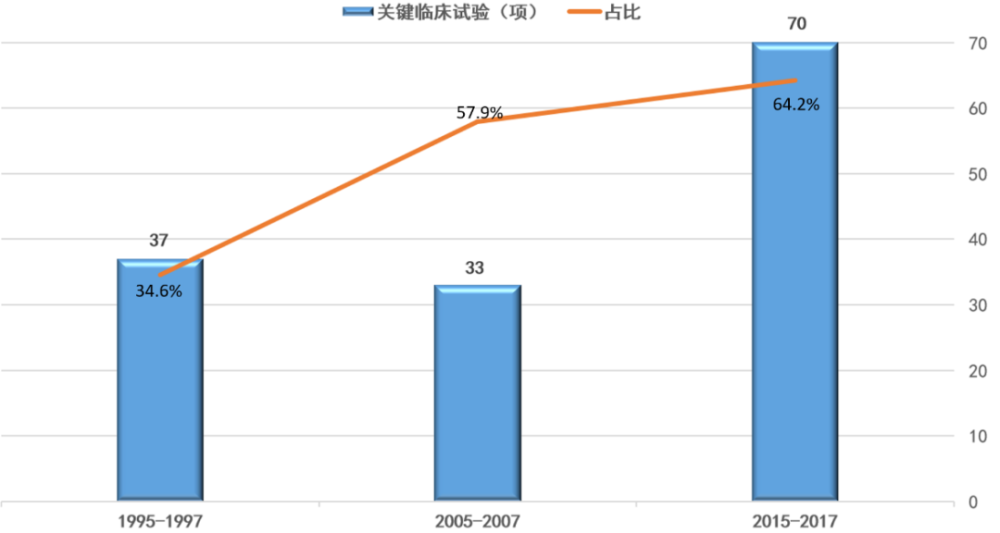
▲采用至少一项加快审评审批计划的获批新药、生物制品与占比(数据来源:参考资料[2],药明康德内容团队制图)
采用至少一项加快审评审批计划的获批新药、生物制品的占比,从1995-1997年间的34.6%,增加到2005-2007年间的57.9%,并进一步增加至2015-2017年间的64.2%。获批新药与生物制品所依据的关键性临床试验所涉及的治疗领域,1995-1997年期间最多为传染性疾病,合计53项(占33.8%);2015-2017期间为癌症,合计32项(占27.1%)。
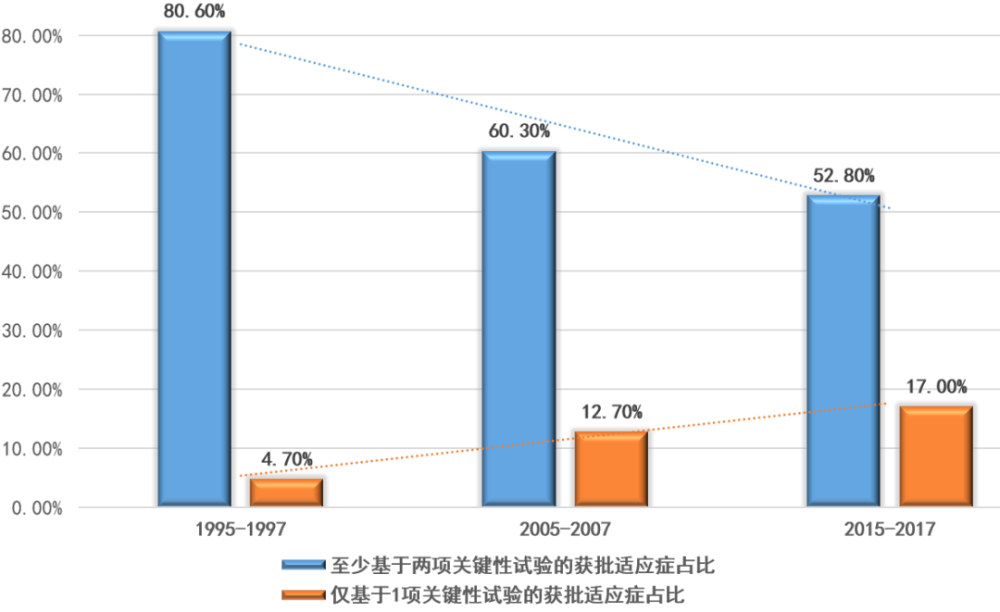
▲至少基于两项关键性试验的获批适应症与仅基于1项关键性试验的获批适应症占比变化趋势(数据来源:参考资料[2],药明康德内容团队制图)
只基于1项关键性试验获批的适应症占比增加,从1995-1997年间的4.7%,上升至2005-2007年之间的12.7%,并继续增加至2015-2017年之间的17.0%。而至少有两项关键性试验支持的获批适应症,所占比例从1995-1997年之间的80.6%,降至2005-2007年间的60.3%,2015-2017年期间进一步降至52.8%。也就是说,在过去20年中,获得FDA批准的新药与生物制品,基于至少两项关键性临床试验占比,下降了近30%。
值得注意的是,获批的适应症中,基于至少一项试验时长为6个月的关键性试验的占比,从1995-1997年之间的25.8%,增加至2005-2007年之间的34.9%,并继续增加至2015-2017年之间的46.2%,20年来的上升幅度超过20%。
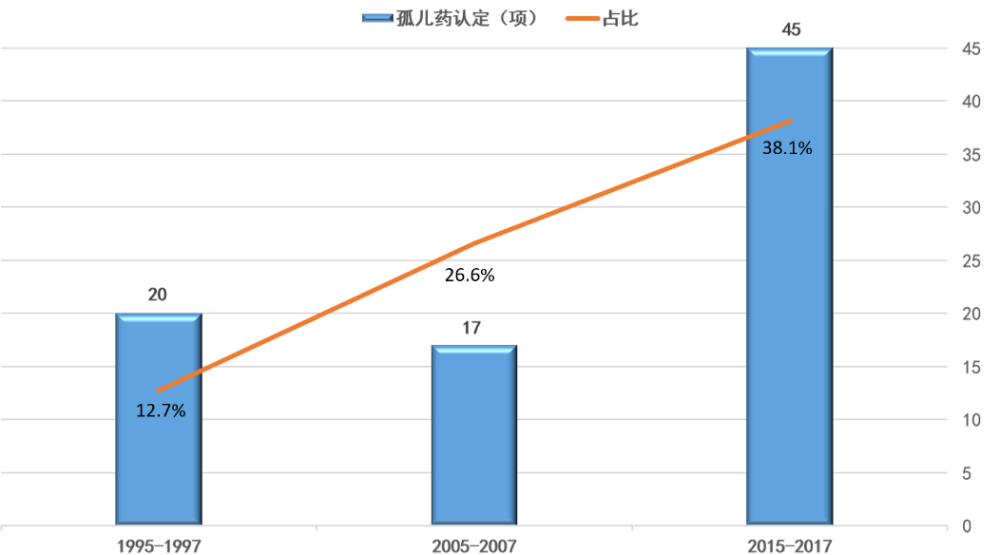
▲获批孤儿药变化趋势(数据来源:参考资料[2],药明康德内容团队制图)
在过去20年中,获批新药与生物制品,基于至少两项关键性临床试验占比下降了近30%。出现显著下降的原因,除了获批新药和生物制品中,孤儿药所占比例显著增加之外,出现下降的背景,是新药研发所针对的疾病领域,从高血压、传染病等,逐渐向癌症转移。近年来兴起的个体化医疗,在癌症治疗药物开发策略方面,也体现出“分而治之,聚而歼之”(Divide and Conquer:Orphanizing All Diseases)的变化发展趋势。
作者的分析还表明,获批的新药和生物制品,朝着针对较窄的适应症治疗药物转向,更多专注于加快上市速度。在调研的时间段内,随着时间的推移,随机化或双盲关键性临床试验占比下降,这样的趋势仍然在持续。分析结果还表明,申办方逐渐从临床终点转向替代终点。
不同国家或地区的药品监管机构,在新药审评审批中,对关键性临床试验的要求,存在差异。例如,日本厚生劳动省(WHLW)属下的药品和医疗器械总合机构(Pharmaceuticals and Medical Divices Agency,PMDA),并没有全盘照搬FDA的做法。FDA和欧洲药品管理局(EMA)通常要求开展两项3期临床试验,来证明相关药品的有效性,但PMDA通常只要求在随机化、设立对照的3期临床试验试验中,只安排一项关键性试验。但在其它方面,PMDA对各期临床试验施加了更为严格的要求。尽管FDA和EMA通常要求申办方在3期临床试验中,证明药品的疗效优于安慰剂,但PMDA通常不强制要求设立安慰剂对照。因此,PDMA对疗效的判断,更多是基于与对照药品相比较。
这项研究的结果表明,在过去的30年中,对于支持新药和生物制品获批的临床试验证据的要求,已经发生了质的变化,更少依据关键性试验,对设立对照的要求有所减低,所需的关键性试验时间更长。这种变化,对临床医生和患者考虑是否使用新获批药品会有影响,原因在于,在新药和生物制品获批所依据的关键性临床试验证据出现变化的同时,对批准后药品的安全性与疗效的要求也不断增加。
四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..四川省医药保化品质量管理协会举办2026
当前,制药行业正处于合规升级与绿..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..四川省医药保化品质量管理协会党支部召
按照省市场监督管理局社会组织联合..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


