

依据美国法律要求,新药在获得批准前进行测试,确保药品能够提供与风险相称的明确的获益。FDA面临的一个重大挑战,是如何在严格检测,与及时批准利大于弊的药物之间,取得恰当的平衡。1983年至今,FDA在审评审批方面的改革,成就有目共睹。回眸35年来在药品审评审批方面的变化发展,促使大家思考,百尺竿头,如何更进一步。
近日,哈佛大学医学院布列根妇女医院医学系监管、治疗药物与立法计划Jonathan J. Darrow博士等,在《美国医学会杂志》( Journal Of The American Medical Association,JAMA )发表论文,详细回顾和阐述了1980年至今,FDA在药品审评审批方面的进展,讨论了进一步改革。
通过相关立法,优化审评资源
1962年《Kefauver-Harris修正案》颁布之后,FDA对临床证据提出了更高的要求。对美国新药价值的信心,以及美国处方药报销的环境,使得美国的医药产业成为全球最大的行业之一。但与此同时,药品审评迟滞(drug lag)备受诟病。医药商、患者权益团体和来自于各方面的人士认为,药品获批所需的时间过长,推高了药品研发成本。各界呼吁,通过壮大FDA审评队伍,同时适当放松批准前标准,增加激励,消除药品审评迟滞,减少药品审批时间,加快新药上市。
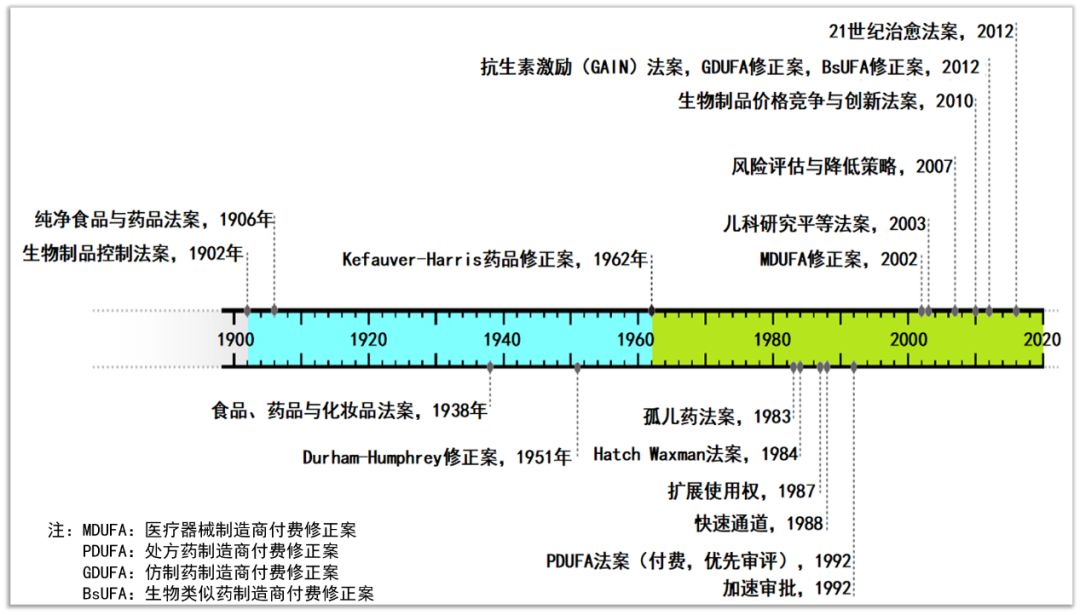
▲1902年以来,FDA药品审评审批相关的立法发展变化情况(药明康德内容团队制图)
从1983年到2018年,通过立法贯彻执行相关法规,FDA的药品审评审批,成就有目共睹。 从各个阶段的新药获批数量看,获批的新药和生物制品许可,实现显著增长。1990-1999年,包括生物制品在内的新药平均批准数量为34个/年,2000-2009年为25个/年,2010-2018年为41个;生物制品许可,从1990-1999年的中位数2.5个,2000-2013年的5个,增加到2014-2018年间的12个。简化新药(仿制药)审评审批方面,从1970年到1984年《药品价格竞争与专利补偿法案》(简称为Hatch-Waxman法案)颁布,每年批准的仿制药品中位数为136个;从1985年到2012年《仿制药生产商付费法案》颁布,每年批准的仿制药中位数为284个。开始实施《仿制药生产商付费法案》后的2013-2018年,每年批准的仿制药中位数为588个。处方药使用者费用的资金从1992年的新药和生物制品,在2012年扩展至的仿制药和生物类似药。截止于2019年11月18日,FDA已经批准了25个生物类似药。
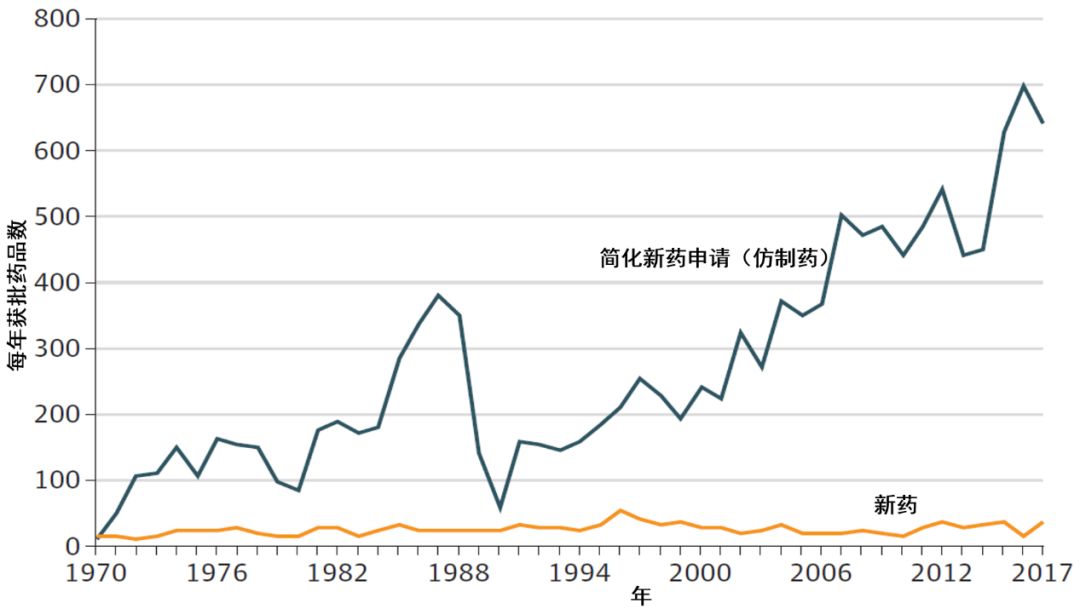
▲1970-2018年,FDA药品批准情况( 图片来源:参考资料[6])
1992年颁布《处方药使用者费用法》,依据该法案的处方药生产商付费总额,从1993年至1997年的年均6600万美元,增加到2013-2017年之间的年均8.2亿美元。2018年,PDUFA付费总额,约占同期支付给新药审评雇员薪金总额的80%。2017财年,FDA用于人用药品与生物制品的总支出为15.5亿美元,其中的12.2亿美元来自于相关的付费,占总支出的79%。
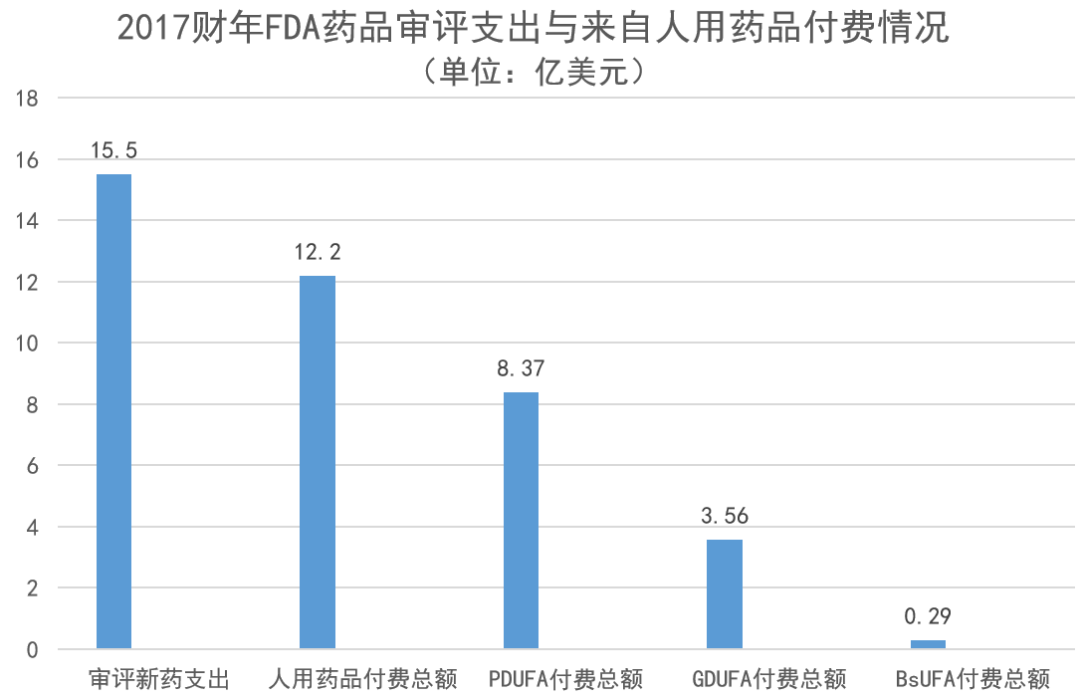
数据来源: 参考资料[1], [2],药明康德微信团队制图
多管齐下,理顺审评通道
依据《孤儿药法案》认定获批的药品占比,从1984-1995年之间的18%(55/304),增长到1996-2007年之间的22%(82/379),2008-2018年间,这一比例继续增长至41%(154/380)。除了孤儿药认定之外,还通过多个立法,建立了多个有针对性的加快审评审批计划。
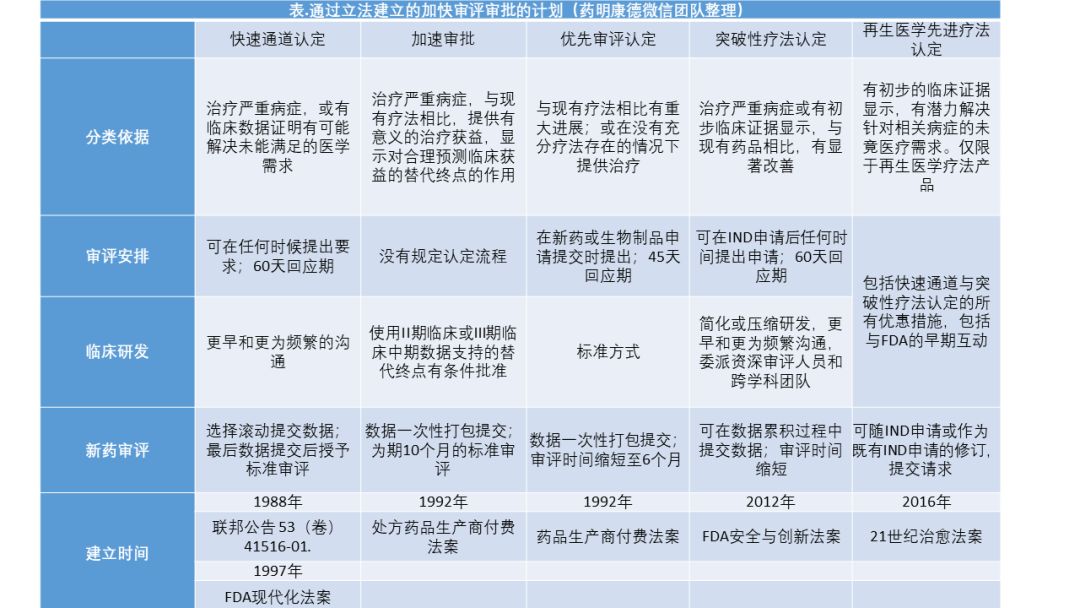
▲通 过立法建立的加快审评审批的计划(药明康德内容团队制图),点击可见大图
随着时间的推移,FDA在新药审评审批中,使用加速批准,快速通道和优先审查的数量有所增加。2018年,有81%(48/59)的获批新药,受益于这类计划中的至少一项。至少依据2项关键性试验获批的新药,从1995-1997年间,占124个获批药品中的80.6%,下降至2015-2017年间,占106个获批药品的52.8%;而参加相关临床试验的患者中位数,并没有显著变化(774名 vs. 816名)。
审评时间显著缩短
备受各界关注的FDA药品审评时间,从1983年的>3年,减少至2017年的<1年;但是,同期从临床试验申请获得默认准许,到药品获批的总时间,却一直维持在8年左右。
标准申请和优先申请(包括所有审评周期)的审评中位时间,从1986-1992年间的2.8年,下降到1993-2005年间的1.5年,2006-2017年间的1.2年。2018年,标准申请的中位审评时间为10.1个月(n=79),优先申请的中期审查时间为7.6个月(n=43)。
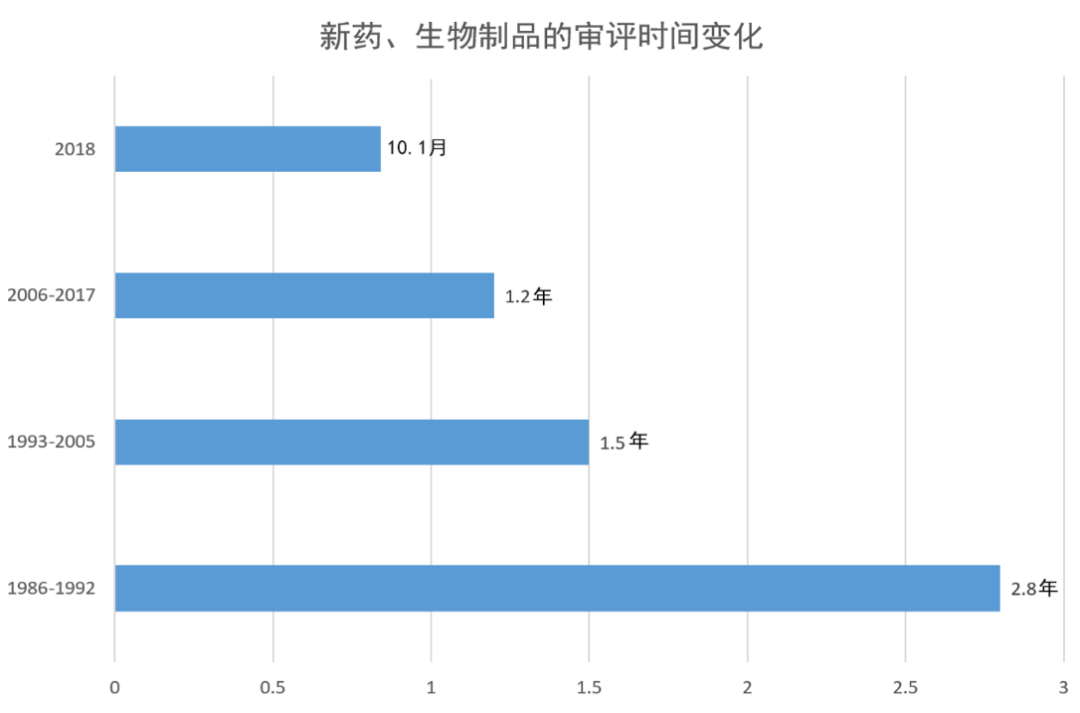
数据来源:参考资料[6],药明康德内容团队制图
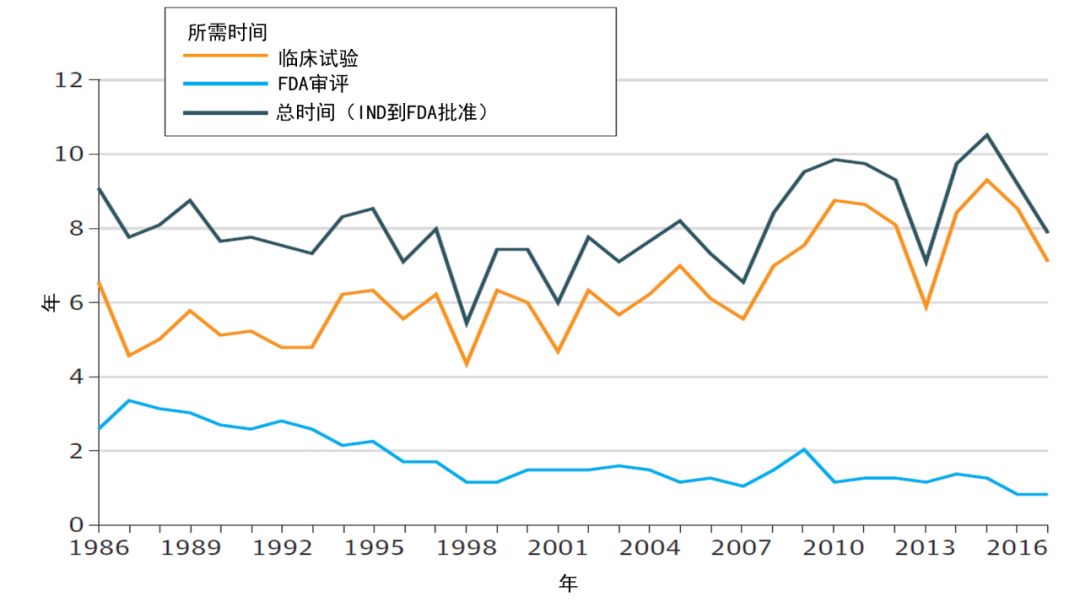
图片来源:参考资料[6]
在2015年至2018年期间,获批的172个药品中(包括标准审评与优先审评),首轮批准率为90%;而此前的2011年至2014年期间,获批的137个药品中,首轮批准率自1988年以来,在提交的新药申请与生物制品许可申请中,获批率约为80%或更多。FDA的内部审评时间已经缩短,与其它国家或地区的药品监管机构的审评时间相当,或更低。但是,从新药临床试验(IND)获批到药品最终获批所需时间,未减反增。
“灵活”应对
1983年《孤儿药法案》确认,"对孤儿(罕见病)药物的临床检测,不能在与对普通疾病的药物检测相同的要求下进行。FDA对罕见病用药的上市前测试的灵活要求,引起了各界对基于非随机试验证据基础的关注。始于1988年的快速通道,使得只根据2期临床试验(即关键性临床试验)证据,就批准临床亟需的药品成为可能。1992年,依据PDUFA法案开始实施的“加速审批”计划,使药品依据替代终点(surrogate endpoints)获批成为可能。通过“加速审批”计划,可以不依据临床终点本身获得改善为依据,而是依据“有可能预测临床获益的”替代终点。但是,通过加速审批的药品,上市后必须完成批准后研究,证实相关药品的实际临床获益;否则,依法加速撤销批准。2012年颁布的《FDA安全与创新法案》,增加了突破性治疗药物计划,该计划的功能和意图,与快速通道计划相似,除了药品监管方提前介入之外,内部审评流程更加规范2016年颁布的《21世纪治愈法案》(The 21st Century Cures Act),建议在授权FDA在批准新药时,通过鼓励使用影像学或实验室研究,形成确定功效的依据,对批准标准产生深远影响。

▲1988年,FDA公布加速针对无药可治的患者的新治疗药品审评,被视为“快速通道”的开端 (数据来源:参考资料[13],药明康德内容团队制图)
这些临床试验生成的证据,被添加到临床前信息中,并作为新药申请(NDA)的一部分,提交给FDA;FDA评估这些试验是否支持医药商所宣称的疗效,并证明该药物足够安全。相关法律规定的“充分和严格合规”(adequate and well-controlled)试验支持疗效说明的要求,变得更加灵活,同时也更具争议性。例如,任何一项试验都可能受到未被发现的系统偏见的影响,因此,1962年的药品批准法规,一般需要2次充分和严格合规的随机调查,但1997年国会将依据“单个临床试验”(single clinical trial)的“非正式”做法写入了立法。在某些情况下接受单个临床试验,例如,在有来自于当前研究人群以外的人群的数据提供额外支持的情况下,或者在设计周密的多中心研究提供高度可靠和统计学上强有力的证据的情况下。
孤儿药:占比将会更高
依据《孤儿药法案》被认定为孤儿药的获批新药比例,从1984-1995年的18%(55/304),1996-2007年的22%(82/379),2008-2018年增加到41%(154/380)。
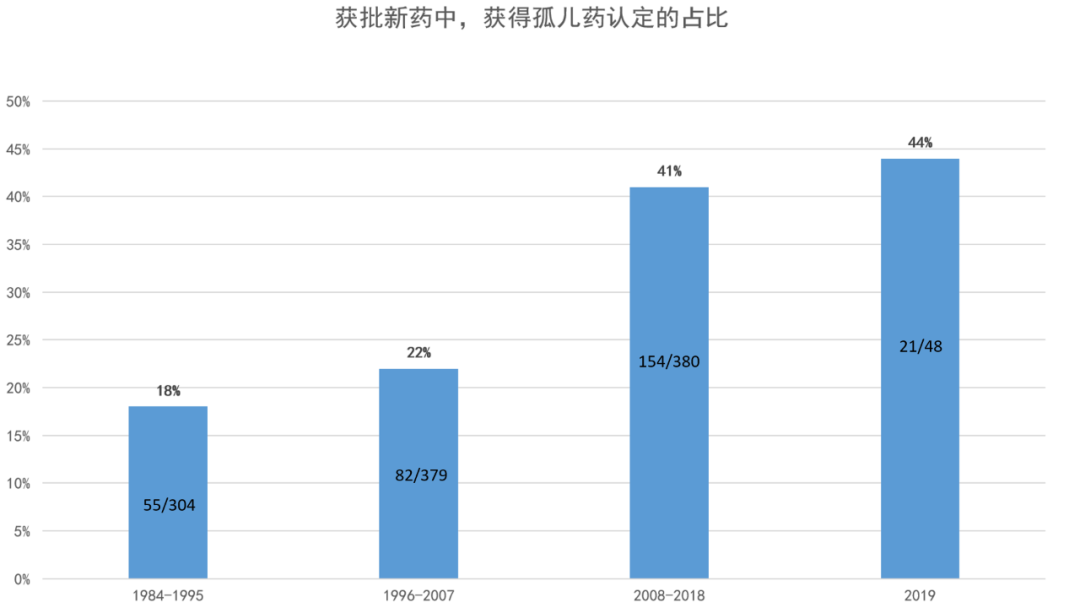
数据来源:参考资料[6], [18],药明康德内容团队制图
与用于治疗常见疾病的药物试验相比,孤儿药的临床试验规模较小,临床试验随机化或双盲的可能性较小,评价疾病响应的情况更多,但评价总生存期的情况更少(参见下图)。由于基因组学研究的进展,按照基因组差别,将常见疾病划分为不同亚型的情况越来越多,尤其在肿瘤学领域。因此,预期罕见病用药在获批新药中的占比,还会继续增加。
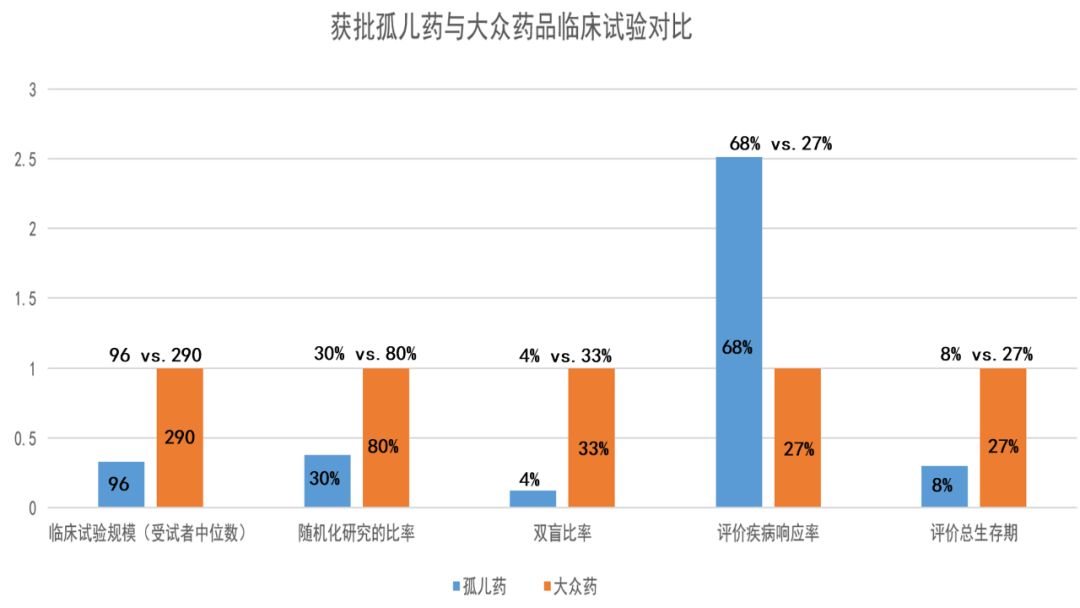
数据来源:参考资料[19],药明康德内容团队制图
突破性疗法
2013年底,Gazyva (obinutuzumab)获批用于慢性淋巴细胞白血病治疗,成为首个获批的突破性疗法。2014-2019年间,获批的新药中,27%被授予突破性治疗药物认定(70/261)。获批的突破性治疗药物认定的药品,研发时间显著缩短(4.8 vs. 8年)。
2018年,来自于相同研究机构的Darrow等,在《新英格兰医学杂志》( NEJM )上发表的研究报道,2013到2016年期间,在所有获批的突破性疗法认定的药品中,52%(16/31)依据1期或2期临床试验获批;54%(14/31)依据单个试验结果获批,42%(13/31)的临床试验没有采用活性对照或空白对照。这种情况,对癌症用药尤其显著。
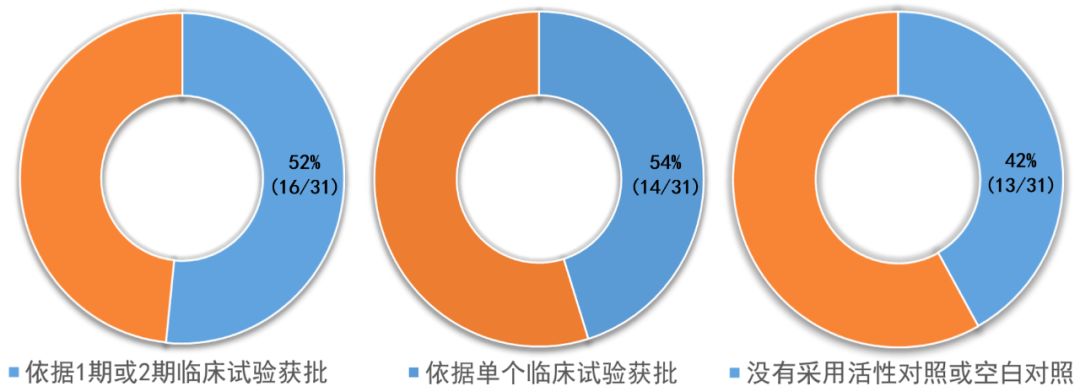
数据来源:参考资料[6],药明康德内容团队制图
Hwang等2016年发表于 Journal of Clinical Oncology 上的一项研究结果,对2012年至2017年间获批的33个获得突破性疗法认定的癌症用药,以及同期获批的25份没有获得突破性疗法认定的癌症用药的分析表明,对于实体瘤而言,获得突破性疗法认定的新药与没有获得突破性疗法认定的新药相比,在响应率(response rate)(37% vs. 39%),死亡率(6% vs. 4%),以及严重不良事件(38% vs. 36%)方面,两者间不存在差异。
药品批准趋势
1986-1996年间,48%的药品(150/313)具有至少一项加速计划资质,1997-2007年间,这一比率为51%(163/319),而2008-2018年间,为64%(243/380)。
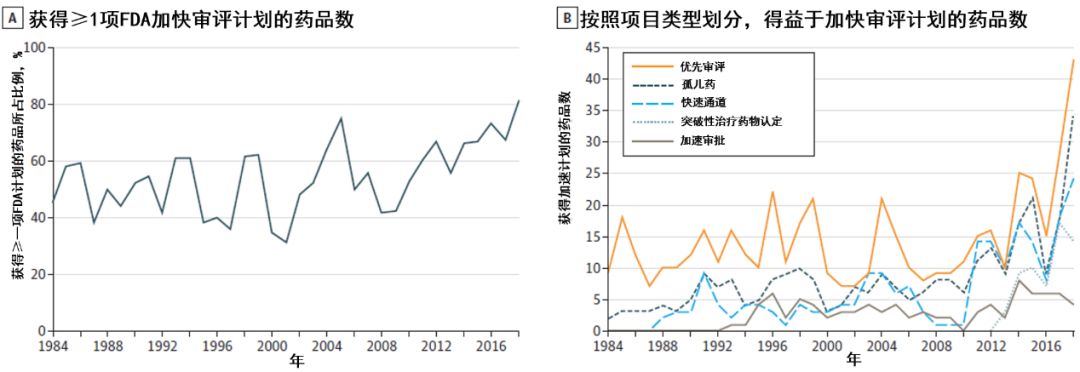
图片来源:参考资料[6]
自1986年以来,新药(包括生物制品)的年平均批准数,1990-1999年期间为34个,2000-2009年期间为25个,2010-2018年期间为41个。1986-1996年,CDER批准了21个治疗用生物制品,1997-2007年批准了44个,2008-2018年批准了88个。1998年至2018年批准了42个疫苗,1998-2008年间,平均每年批准2.0个,2009-2018年间,平均每年批准1.8个。
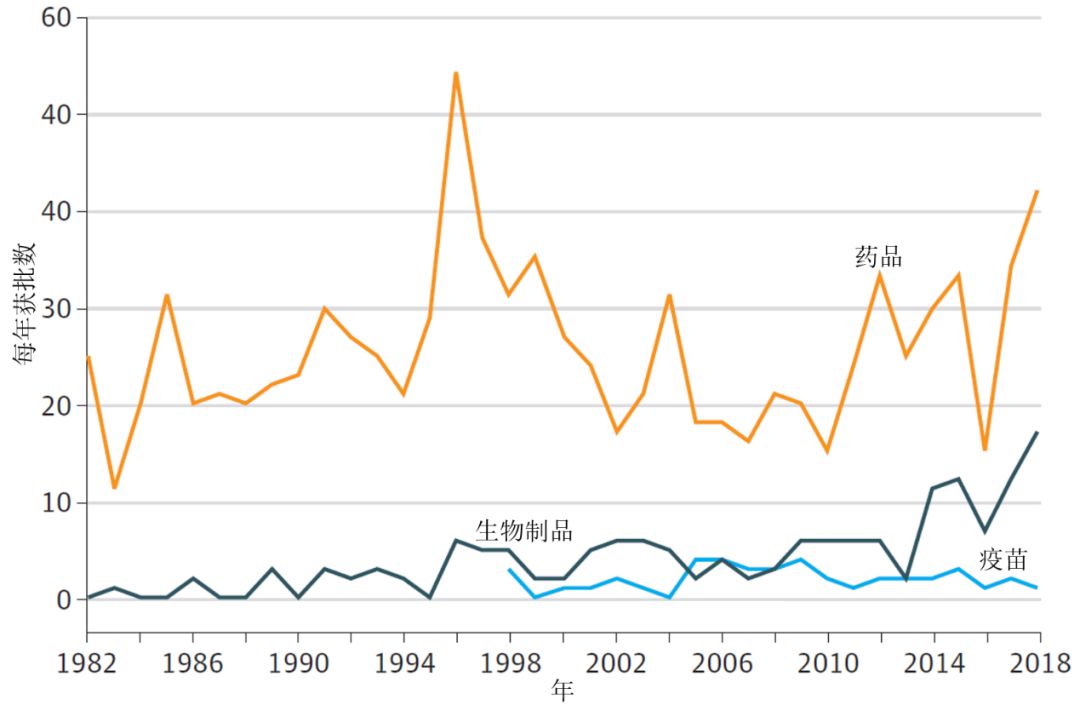
图片来源:参考资料[6]
允许依据较少或规模更小临床试验,以及早期临床试验,批准新药,有助于加快药品上市速度,降低新药临床试验成本,显著降低管理成本。相关的临床试验,和随机化、设立对照、置盲的常规临床试验,或基于对患者感觉、机能或生存方式的传统衡量方式,有所区别。此外,相关法律规定的获批标准,仅要求新药“具有其声称或表示具有的效果”,一般不要求药物超过除零以外的任何特定疗效阈值。
药品审批中,替代终点运用越来越多,包括成熟的替代终点。2005-2012年间,获批新药所依据的关键疗效试验中(n=448),44.3%是基于替代终点;2015-2017年间获批的新药中(n=253),为59.3%。
百尺竿头,如何更进一步
尽管FDA的审评时间呈现出显著的下降趋势,但临床开发的总时间并未缩短。在一定程度上,获批新药的临床开发总时间没有显著下降,存在多种原因。例如,在获批新药中,孤儿药的占比上升,但孤儿药临床试验难以招募受试者;此外,诸如中枢神经系统疾病治疗药物,部分早期确认的非常重要的癌症用药,这些相关药品的疗效确认,需要建立更长的时间标准。1962年《Kefauver-Harris修正案》颁布之后,FDA对临床数据提出的更高要求,使得在全球范围内,FDA的新药审批赢得了尊敬。但是,由于药品监管标准和审批所依赖的证据变得更少,在最终确定药品的获益-风险特征之前,在让患者更快获得新药,以及可能将患者暴露于更多风险之间,需要做出更多权衡。当临床上实际用药的患者情况,与参与临床试验的患者不同时,这些问题可能会加剧。尽管新的治疗药物,为患者提供了额外的选择。但是,如果新药只是根据有限的证据获批,在实际使用中,如果相关药品用于临床获益证据较少的患者,或用于取代那些有明确证据,效用相当或效用更为确凿的药品,可能会引发问题。由于新的治疗药物,往往比旧的治疗药物更昂贵,这些缺陷,可能会使得相关的新药,做不到真正的物有所值。
在药品开发速度与严格的证据生成之间,实现适当的平衡,需要构建周密的监管要求,正确执行相关法规,并根据证据,来评估相关的审评计划,是否真正能够使公众获益。尽管从上世纪80年代以来,实施的许多加快审评计划,取得了显著成功,在改进审评流程的同时,使得患者能够更早获得相关的药品,但无可否认,其中的一些计划,也存在临床结局没有得到充分表征的潜在缺陷。国会、行业应定期重新评估这些后果之间的平衡。作者认为,还需要更多证据,说明这些计划,在多大程度上,可能带来得不偿失的行政负担,没有真正做到改善研发产出率,或者在对FDA的有限资源造成竞争。甚至导致了将注意力放在更容易测量,但相关性较低的指标上,例如批准速度,或是每年批准新药的数量方面。检验药物审批架构是否成功,最终取决于相关药品对患者福祉的贡献或减损的程度,包括对用药成本的影响。理想情况下,监管与激励架构,应基于真正与患者相关的治疗获益的可量化措施,这些措施,应该能够明确、客观地传达。
在同一期的 JAMA 中,登载了约翰-霍普金斯大学Bloomberg公共卫生学院教授,曾经担任FDA副局长的Joshua M. Sharfstein医生署名的社论。
Sharfstein教授认为,随着时间的推移,FDA的相关监管流程,演变发展为成了一系列具有灵活审评标准和激励措施的有针对性的计划。为了进一步发挥药品审批制度带来的获益和价值,进一步降低药品审批体系的低效率和风险,有必要进行FDA改革。Sharfstein教授建议,从4个方面着手。首先,国会和FDA,应该使各种快速审评计划进一步合理化。其次,对于具有重大获益但可能带来严重风险的药品,FDA应该着力加强相关药品上市后安全性监管。第三,国会应该重新调整为相关医药公司提供的具体的上市销售保护计划。第四,国会应该应用专利和定价激励措施,促进加速审批药品的最终证据的生成。
四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..四川省医药保化品质量管理协会举办2026
当前,制药行业正处于合规升级与绿..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..四川省医药保化品质量管理协会党支部召
按照省市场监督管理局社会组织联合..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


