

4月23日,FDA合规办公室(OC)发布《2025年合规办公室年度报告》(本该去年发布的2024年报被跳过),概述了该办公室在药品生产设施合规监管、临床试验监管、生物研究监测等方面的工作成果。报告显示,FDA在2025年的合规行动显著增加,特别是针对未经批准或错标的药品违规行为以及药品掺杂违规行为的监管执法力度加强。
总体数据
根据报告,合规办公室在2025年共发出314封警告信,较往年显著增加(2023年报中的数据是170)。此外,还有321起药品召回事件,涉及755个产品;与企业举行了85次监管会议;从FDA的药品注册和清单系统中停用了10922个药品。
在合规审查方面,合规办公室发出了970封药品生产检查分类信(2023年仅为218);为潜在的ClinicalTrials.gov违规行为发出了42份初步违规通知;发出了8700多份药品产品的电子证书,证明设施符合FDA标准以支持出口;与外国监管机构共享了409份检查和合规文件。
警告信:聚焦生产CGMP和未经批准药品
报告显示警告信类型分布:药品生产设施CGMP/掺杂违规32%(约100封),未经批准或错标药品违规20%,配药虚假宣传19%,互联网药店相关违规12%,未经批准或错标药品和CGMP/掺杂违规7%,临床研究/机构审查委员会违规4%,配药设施违规4%(见下图)。
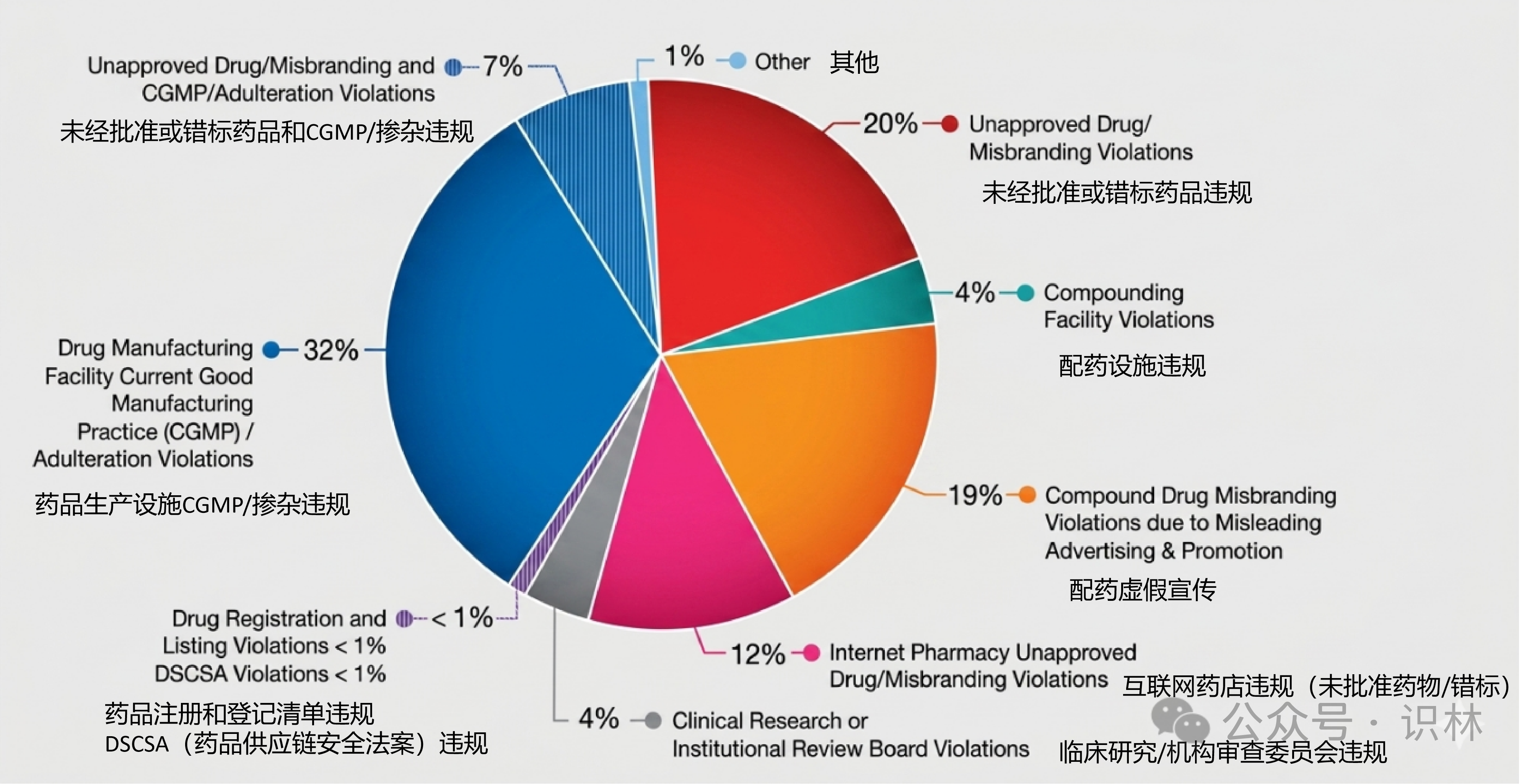
*原图较模糊,上图为AI转制高清图片,翻译供参考
驱动警告信增长的不止是我国药企关注的生产现场检查。除了两个主要领域:未经批准或错标药品违规(180多封),以及药品掺杂违规包括CGMP偏离(120多封),虚假或误导性广告导致的配药错标(19%)是2025年警告信大幅增加的最大单一驱动因素。这一点与2025年FDA发起打击虚假广告行动有关。
生产设施监管:970封检查分类信,125封工厂警告信,76家进口禁令
FDA通过监督CGMP要求帮助确保美国药物安全有效。所有为美国市场生产的药物必须满足相同质量标准,无论产地。
FDA审查所有发出483表的药品生产设施检查报告并分类,包括NAI(无行动指示),VAI(自愿行动指示),OAI(官方行动指示)。2024年10月,FDA机构重组实施新合规模式,将VAI检查分类集中至CDER合规办公室,取代先前基于地区的审查系统。合规办公室继续处理OAI检查分类。2025年发出970封检查分类信,96%在90天目标内完成。这个数据看起来高效,但从行业反馈来看,这个分类应该还包括所谓的pOAI(潜在官方行动指示)。该分类为FDA提供较大灵活性,但也为药品审批带来不确定性。
合规行动方面,合规办公室向药品生产商发出超过125封警告信(其中15封基于第704(a)(4)节的记录索要,显示FDA持续使用替代监管工具)。将76家公司添加到CGMP相关进口禁令,其中55家的进口禁令由替代监管工具驱动。
合规办公室还参与了多项指南制定。2025年的重点文件包括:《GDUFA 下的警告信后会议》,《遵循21 CFR 211.110的考虑》,《医用气体CGMP》修订草案,《进行远程监管评估 - 问答》。
其中报告还特别强调扩大了远程监管评估(RRA)的使用范围,包括依据第704(a)(4)节提出的强制性记录索要,以快速获取信息并提高检查效率。当企业未能充分回应时,FDA可发出警告信,或将其置于进口禁令。
临床试验监管:评估1000多起投诉,141份临床检查摘要
通过生物研究监测(BIMO)计划,合规办公室在2025年评估了1000多起临床试验相关投诉和举报,进行了超过590次检查和RRA,涉及申办者、合同研究组织、临床研究员、机构审查委员会。合规办公室还针对上市后不良事件报告要求和风险评估和减轻策略(REMS)要求进行了超过55次检查和RRA。
2025年,合规办公室向新药办公室提供了141份临床检查摘要以支持上市申请审评。发出14封警告信(11封给临床研究员或申办者-研究员,3封给申办者)和6封无标题信。关于GCP检查,还可见FDA的分析文章。
报告还称,在ClinicalTrials.gov监管方面,发出42份初步违规通知信和2份违规通知信,此后近90%责任方成功发布所需临床试验信息。但这个信息跟严重的违规情况并不相称。不久前FDA发出2200封信函敦促相关方合规登记临床信息。
关于举办中药饮片炮制技术研究与质量控
各会员单位: 为顺应药品监管规..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


