

制药企业进行验证活动时——可能会出现测试结果与验证目标不一致的情况,对于同一个不一致,不同的原因处理方式亦不同。对偏差进行分类管理,以不同的方法和流程区分对待,一方面能达到降低质量风险的目的,另一方面企业的偏差管理体系会更加系统、科学、高效。
根据各国GMP相关法规和指南的要求,制药企业应当有计划地开展验证活动,以保证整个生产过程受控。而验证过程中会不可避免地出现与验证目标不一致的情况。对于这种“不一致”,只有基于一套完善的偏差管理体系来支撑,才能真正达到准确处理与风险控制的效果。
验证过程中的偏差发生
偏差系指偏离已批准的程序(指导文件)或标准的任何情况(引自ICH Q7,原文为:Deviation - Departure from an approved instruction or established standard.)。因此偏差发生的前提是已经预先定义了规则,偏差管理的基础是有效的,足以控制生产过程和药品质量的程序(指导文件)或标准。
验证活动是按照被批准的验证方案来实施的,在这份验证方案中,必须清楚地界定关键系统属性和参数,以及相关的可接受标准。在验证执行过程中,任何与可接受标准的偏离,都属于偏差的范畴,如未按照方案执行,出现了不符合既定可接受标准的结果等。对于出现的偏差,必须真实地记录在报告中,并根据偏差管理的程序作出对应的处理,最终在所有的偏差已被关闭或者其影响可接受时给出验证成功的结论。
验证过程中的偏差分类
我们应该注意到,验证本身即是证明预先提出的程序或标准是否可以被确认的一个过程,即使导出了与预期不一致的结果,也不能说明验证对象(设施设备、仪器或系统)会对产品质量等造成影响(无论影响大小)。验证过程本身也是一个不断完善的过程,是“提出标准或标准程序——实施验证或确认——得出结论——重新审视标准——验证对象修改或预期标准调整——再次验证或确认”的一个螺旋前进或上升的过程。
具体来说,验证是证明任何操作规程(或方法)、生产工艺或系统能够达到预期结果的一系列活动。即验证是在证明方法或者程序是适用的,是通过先设立一个理论上的标准,在经由一系列验证活动,最终建立或确定标准的过程。这个过程区别于正常生产的状态,正常生产中的标准是确定的或者是已被验证的。而验证过程中的“标准”是一个预设的标准,是正常生产标准的初始值、理论值或经验值,经过验证之后,这个“标准”才被最终确定为正常生产的标准。
在验证过程中产生的偏差,一部分是对已确定标准的偏离,一部分是对正在建立的标准的偏离。已确定的标准,包括但不限于已批准的操作规程、验证所用仪器设备、对物料的标准、对环境的要求等。正在建立的标准,比如设备本身未经过确认的属性(已经过确认的属性为已确定的标准)、设备运行的方法与能力、清洁设备的方法等。对这两种不同标准的偏离,将验证过程中出现的偏差分为了两类:常规偏差和验证偏差。
换言之,常规偏差是对已确定标准的偏离,除了验证过程中的某些情况,主要发生在正常生产过程中,产生于已验证的对象,如清晰明确的生产工艺、物料平衡限度、质量标准、检验方法、操作规程等。
尽管任何企业都无可避免地在生产过程中产生偏差,但这类偏差理论上是不应该发生的,制药企业应通过完善的组织机构、合理的文件系统和充分的人员培训来最大限度地预防偏差的发生。正如《药品生产质量管理规范(2010年修订)》中的要求:“各部门负责人应确保所有人员正确执行生产工艺、质量标准、检验方法和操作规程,防止偏差的产生。”而验证时发生的常规偏差,GMP附录《确认与验证》中亦有明确的要求:“企业应当在报告中对确认与验证过程中出现的偏差进行评估,必要时进行彻底调查,并采取相应的纠正措施和预防措施”。
对于验证偏差,是对不确定标准的偏离或不符合。比如设备在进行安装确认(IQ)时,PID图纸与实际情况有差异,此时的图纸是设计的“标准”,不是经过确定的最终标准,因此这种差异是对不确定标准的差异。又如洁净区的更衣程序,验证过程中发现当前的程序不满足微生物限度的要求,由于此时的程序是不确定的,因此造成的不符合要求的结果也属于对不确定标准的偏离(这种不确定标准是双向的,即验证对象的不确定和接受标准的不确定)。GMP附录《确认与验证》中这样描述这类偏差:“当验证结果不符合预先设定的可接受标准时,应当进行记录并分析原因。企业如对原先设定的可接受标准进行调整,需进行科学评估,得出最终的验证结论。”该条款不仅给出了验证偏差的范围,也给出了应采取的行动。
对比GMP中这两类偏差的描述,可见对于常规偏差和验证偏差的管理要求不同,其处理方式也是不同的。
验证偏差与常规偏差的区别
发生的原因不同
验证偏差是由于验证对象本身的不确定因素造成的,因为验证对象未经过确认或验证,不可避免地会出现不符合既定的验证目标值的情况。比如贴标机运行能力的验证,在URS中,贴标机的运行能力需求为不低于250 瓶/min,设备厂家提供的设备说明为300 瓶/min,于是方案中的可接受标准设计为300 瓶/min,而验证时只能达到280 瓶/min,这种偏离的原因显然是方案中的标准是厂商设定的、共性的、理想状态下的目标值,而验证的结果才能真实地反应出这台设备的实际能力。
常规偏差发生的原因很多,更加复杂,并且往往需要调查才能找出根本原因。验证过程中发生的常规偏差,可能是用于验证的方案不适用,比如使用了错误版本的方案;可能是测试所用的仪器不符合条件,比如超出了校准有效期或者在有效期内但是出现了超出计量范围的异常值;可能是物料不满足要求,比如使用了错误批号的物料或者未被放行的物料;可能是人员操作不规范,比如未按照要求取样造成样品受到污染等。这些引发偏差的原因或简单或复杂,或单一或多重,归根结底都是可以规避的,都是“可以不发生”的。
处理方式不同
验证偏差需要记录和解释,必要时采取措施达到一致,如通过科学评估对原先设定的可接受标准进行调整,或者通过调整验证对象使两者一致。比如在“发生的原因不同”一节中贴标机运行能力验证的例子,发现运行能力达不到目标值,先记录偏差,再分析原因是设备本身的属性达不到验证目标,需要采取的行动是首先评估280 瓶/min是可以满足车间对生产能力要求的,因此将验证方案中的300 瓶/min的标准重新定义为280 瓶/min。此时的280 瓶/min是经过验证的标准,当设备投入生产后再出现达不到生产能力的情况时,该偏差便属于常规偏差,是偏离了已确定标准的偏差,处理方式需按照常规偏差的管理程序进行。
常规偏差首先要对其影响性做出初步判断,根据其对质量的影响进行分类,如归为关键偏差、中等偏差或是微小偏差。对于微小偏差,比如在使用之前发现仓库发送了错误的物料,仅需要记录和解释,并且及时更换物料修正错误。对于中等偏差和关键偏差,应调查其发生的根本原因,再评估偏差造成的影响,并建立纠正和预防措施(CAPA),批准后完成纠正行动,最后完成偏差报告。
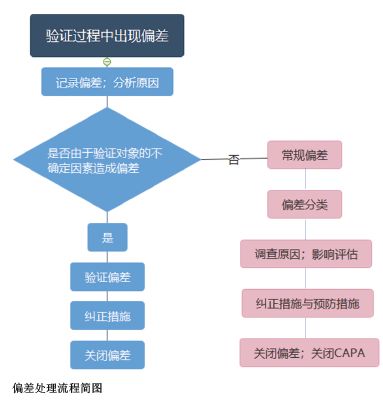
对于常规偏差的处理,需要一套完整的偏差管理体系来支撑,对不同分类的偏差采取对应的处理流程。例如,水系统进行性能确认(PQ)第一阶段时发现某用水点微生物超标,由于第一阶段的水尚未用于生产,因此不需要执行生产产品相关的紧急措施;由于微生物超标会影响未来产品质量,因此该偏差归类为关键偏差; 此时需成立偏差调查小组进行根本原因的调查,并且评估偏差的影响,调查原因和评估影响可采用质量风险管理的方法,利用风险分析工具(如鱼骨图、失效模式影响分析(FMEA)等)得出结论。基于结论,需采取纠正措施来消除偏差的影响,并采取预防措施来避免相同的偏差再次发生以及执行后续的跟踪程序等。
不同类别的偏差处理流程如上图所示。
发生的概率不同
对于任何全新的验证对象,验证偏差为独立事件,只会单次出现,因此验证偏差的处理不需要预防措施。
常规偏差是基于某些原因发生了不该出现的偏离,有可能会再次出现,需在影响评估时分析其发生的可能性,并需要通过预防措施来降低其发生的可能性,从而降低风险,达到风险控制的目的。
关于验证过程中出现的验证偏差和常规偏差,下面将会用两个实例来进行具体分析。
偏差实例分析
实例一
某设备用户需求(URS)中要求其温度精度不大于0.5 ℃,根据厂家文件描述,该设备可控制精度为0.1 ℃,因此验证方案中要求其温度精度不大于0.1 ℃,验证时发现该设备只能达到0.2 ℃的精度。
首先记录偏差,并分析发生偏差的原因。
如果出现偏差的原因是这台设备本身达不到验证目标,该偏差即属于验证偏差,应按照验证偏差的处理方式去处理。此时应采取纠正措施,或者升级验证方案,或者改变设备硬件,最终需使两者一致,进而关闭偏差,完成整个验证过程。
如果出现偏差的原因是温度探头损坏,而又因为超出校准时间所以没有被发现,导致验证结果不准确,即设备实际是可满足方案中的精度要求,但测试结果不满足要求,该偏差即属于常规偏差。此时需对偏差进行分类,再执行对应的流程。纠正措施是维修并立刻重新校准温度探头,并且重新验证;预防措施是对计量管理部门提出提前校准的控制要求,包括升级标准操作规程(SOP)、使用软件定期提醒等。
实例二
进行烘箱或灭菌柜的满载热分布时,出现温度分布不均匀的情况。
首先记录偏差,查找原因。如发现是由于布点时人员操作不规范,探头直接接触到了腔体金属部分,导致导热异常,属于常规偏差,处理方式首先进行影响评估,再现场纠正,重新摆放抬头,使之只能接触装载物而不接触金属腔体,最后提出预防措施,例如对探头摆放进行更为标准化的培训等等。
如果造成偏差的原因是满载时放置的物料太多或者摆放方式不合理,即是由于装载量或装载方式的不确定引起的,属于验证偏差。纠正措施为更改验证方案,使用新的装载量或装载方式重新进行热分布验证。重新验证时或许依然不能达到要求,就需要多次调整多次验证最终确定其装载量与装载方式使其温度分布均匀,才能完成此次验证。
经过验证的装载量和装载方式是已确定的标准,如果设备使用一年后进行再验证时发现满载热分布不均匀,此时的偏差为常规偏差,需按照常规偏差的处理方式进行调查、风险分析、纠正预防措施等。
实例对比结论
常规偏差产生于一个已经合规的系统或设备,理论上这类偏差是不应该发生的,应评估偏差对人员安全、产品、环境、质量体系等的影响,并基于这些影响进行纠正措施(CA)和预防措施(PA),即需要现场第一时间的纠正和长远的预防。其目的在于减少同类偏差发生的概率,降低质量风险。
验证偏差产生于一个未经过验证的系统或设备,这类偏差的处理通常只是陈述事实,并且进行纠正,不需要预防。其目的是如何从偏差状态中恢复,以继续验证的实施。如果对于验证偏差,仍然规定需要实施预防措施,则会占用大量资源,甚至得出不可能完成的行动项。
比如在上文中的“实例二”,装载方式问题,基于该设备是首次引进的新设备,URS只对腔体体积做了要求,且设备满足这一体积要求。如果在下次类似设备验证中保证装载方式不发生产生偏差,则需要的预防措施为:在下次采购前,必须列出该腔体一次使用时所有需要装载的产品或工器具数量、大小、形状,且将上述物品全部在设备制造前就送至设备厂家,由厂家在设计确认(DQ)时就拟定具体的装载方式,且对设备进行非标制造。很明显,上述预防措施几乎是不现实,定制化非标设备的价格也不是标准设备所能比拟的,因此虽然理论上行得通,但需要花费大量时间、人力和费用,并不合适,也没有效率。
总结
验证过程中产生的偏差,一部分属于验证偏差,一部分属于常规偏差。制药企业应当建立完善的偏差管理体系,以区别处理这两类偏差,既能最科学、经济地达到验证目的,又能降低质量风险,实现将风险控制在可接受的程度的目标。
【参考文献】
[1] ICH Q7,Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients,2000.11.
[2] EU Legislation–EudraLex,Volume 4-Guidelines for good manufacturing practices for medicinal products for human and veterinary use,2015.03.
[3] EU Legislation–EudraLex,Volume 4-Guidelines for good manufacturing practices for medicinal products for human and veterinary use,Annex 15 Qualification and Validation,2015.10.
[3] 国家食品药品监督管理总局,药品生产质量管理规范(2010年修订),2011.03.
[4] 国家食品药品监督管理总局,药品生产质量管理规范(2010年修订),附录,确认与验证,2015.12.
[5] 国家食品药品监督管理局药品认证管理中心,药品GMP指南,质量管理体系,2011.08.
[6] 识林网站,IPEM讲义,变更和偏差管理以及CAPA,2018.08,http://lib.shilinx.com.
本文作者就职于杨凌步长制药有限公司。本文由浙江瑞爵制药有限公司运营副总监罗逸寰审核。
关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..关于收取2026年度会费的通知
各会员单位: 在过去的一年里,..四川省医药保化品质量管理协会举办2026
持续提升合规 智慧拓建高质 四川..四川省医药保化品质量管理协会党支部召
按照省市场监督管理局社会组织联合..


