

一、概述
此次仿制药一致性评价是“三医联动”改革的一个重要组成部分,其影响不仅仅限于仿制药,而是对药品质量疗效提出了全新的、更高更严格的标准,对行业进行全面地规范和清理。“一致性”是对“仿制药质量和疗效一致性”的简称,言中之意,“一致性”包含仿制药在质量、疗效两个方面与参比药品一致。药品的质量通常与一系列标准、规范相关,如《中华人民共和国药典》、《药品生产质量管理规范》(GMP)、《药品经营质量管理规范》(GSP)及其他药品质量标准等,通过技术和管理手段保证药品达到这些标准和规范的要求。疗效一致(TE)通常用生物等效(BE)来评估。
二、起始
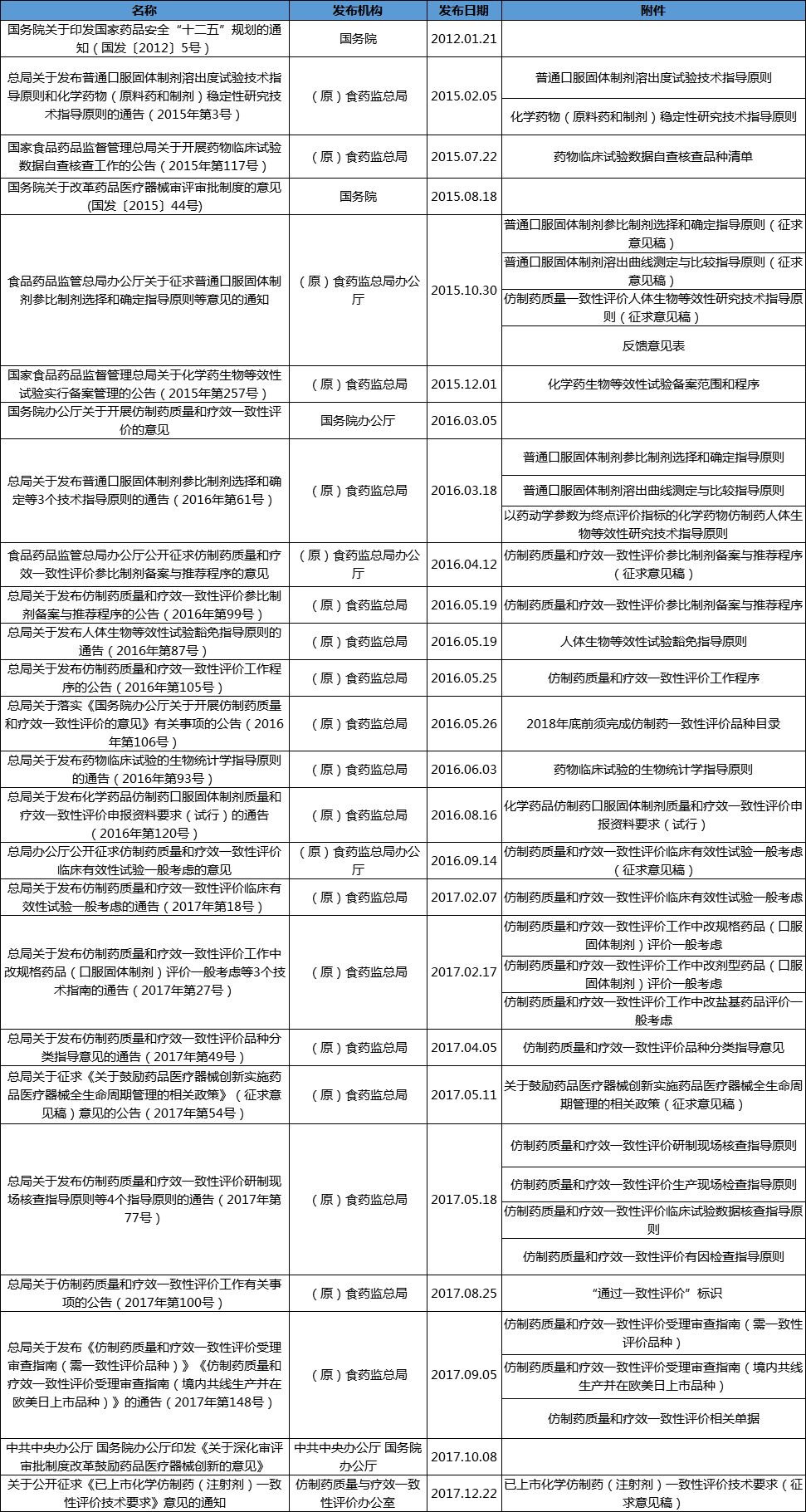
早在2012年1月,《国务院关于印发国家药品安全“十二五”规划的通知(国发〔2012〕5号)》就指出了当时国内药品行业存在的问题,提出了奋斗目标。此后到2016年3月间,原食品药品监督管理总局发布了数个文件,如《总局关于发布普通口服固体制剂溶出度试验技术指导原则和化学药物(原料药和制剂)稳定性研究技术指导原则的通告(2015年第3号)》、《国家食品药品监督管理总局关于化学药生物等效性试验实行备案管理的公告(2015年第257号)》等。期间影响最重大的政策当属《国家食品药品监督管理总局关于开展药物临床试验数据自查核查工作的公告(2015年第117号)》,该文直接引发了被行业内戏称的“722惨案”,也预示着随后进行的一致性评价对各种临床试验数据真实性的严格要求。2016年3月5日发布的《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》标志着本轮一致性评价正式来开帷幕。同月18日,原食药监总局发布了《总局关于发布普通口服固体制剂参比制剂选择和确定等3个技术指导原则的通告(2016年第61号)》,堪称一致性评价中技术指导原则的根本性文件。
三、文件分类
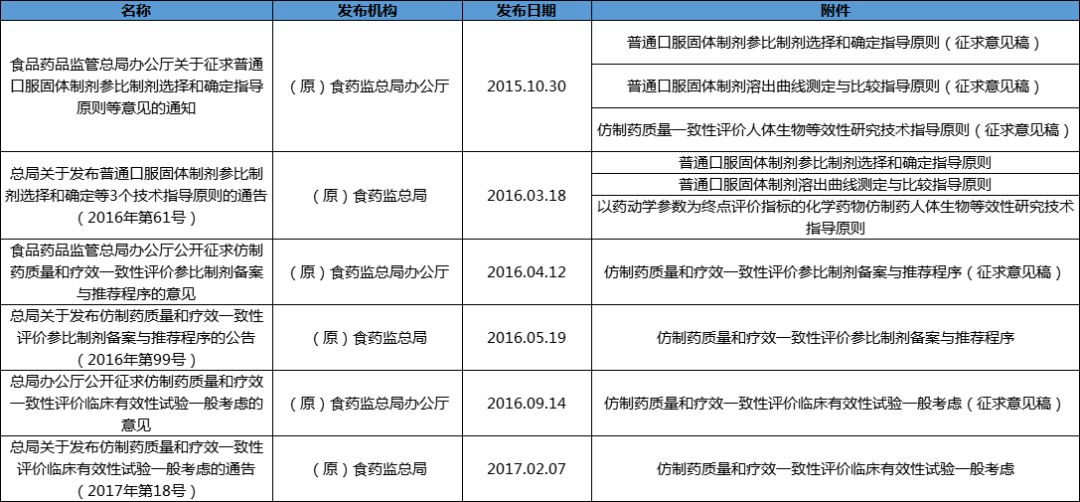
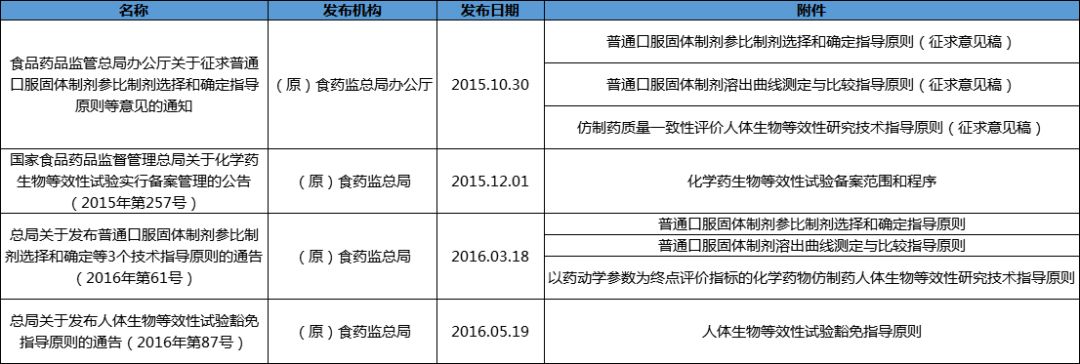
原食药总局2016年第61号通告对参比制剂选定、体外溶出和人体内生物等效性三大方面给出了总体要求。随后发布的各种文件围绕参比备案、没有或无法确定参比制剂的情况、体外溶出研究、体内生物等效性研究、体内生物等效性豁免、现场核查、共线生产品种等方面内容给予详细规定。从剂型分类来看,一致性评价的主体是口服固体制剂。从产品治疗类别来看,应用时间长、临床疗效确切、用量大的品种成为首批要求评价的产品,具体的讲是《总局关于落实有关事项的公告(2016年第106号)》附件中列出289个化学药品种。限于篇幅,此处仅按照参比制剂、生物等效性、口服固体制剂三个分类展示,更多内容见附件。1
参比制剂
生物等效性
3
口服固体制剂
2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..四川省医药保化品质量管理协会举办2026
当前,制药行业正处于合规升级与绿..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..四川省医药保化品质量管理协会党支部召
按照省市场监督管理局社会组织联合..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


